What is it?
Pulmonary Arterial Hypertension (PAH) is high blood pressure in the vessels that carry blood from the heart to the lungs (the pulmonary artery and its branches). By definition, it is diagnosed when the blood pressure in the heart to lung circulation exceeds 25 mm Hg at rest or 30 mm Hg with exercise.
Two types of Pulmonary Hypertension are recognized:
Secondary, in which the cause of the high blood pressure is known; and Primary, in which the cause is unknown.
Primary Pulmonary Hypertension (PPH) is a rare condition, affecting females more frequently than males (2.4 to 1). It is likely a genetic defect and can be inherited.
Secondary Pulmonary Hypertension may be caused by congenital blockages of the pulmonary (heart to lung) circulation. Pressure builds in the pulmonary artery (or its branches) or the pulmonary veins, which carry blood from the lungs to the heart, as the blood backs up because of resistance to blood flow.
Secondary Pulmonary Hypertension may also be caused by late or unrepaired congenital heart disease in which there is a long standing increase in blood flow to the pulmonary arteries. Eventually this results in permanent and progressive stiffening of the pulmonary arteries. The examples of these diseases are atrial septal defect and ventricular septal defect.
Other factors that may result in secondary pulmonary Hypertension include chronic lung disease or sleep apnea which results in low oxygen concentration in the blood (chronic hypoxemia).
How Does Pulmonary Hypertension Develop In Congenital Heart Disease?
The normal heart has four chambers. The two upper chambers, known as atria, are separated from each other by a fibrous partition known as the atrial septum. The two lower chambers are known as ventricles and are separated from each other by the ventricular septum.
Valves connect the atria (left and right) to their respective ventricles. The valves allow for blood to be pumped through the chambers.
Blood travels from the right ventricle through the pulmonary artery to the lungs where it receives oxygen. The blood returns to the heart through pulmonary veins and enters the left ventricle. The left ventricle sends the now oxygen-filled blood into the main artery of the body (aorta).
The aorta sends the blood throughout the body (systemic circulation). The systemic and the pulmonary circulation are separate.
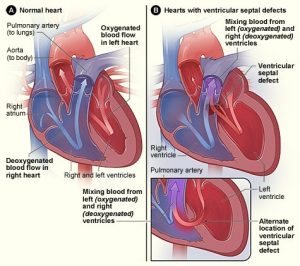
Image from wikipedia
However, some individuals with structural malformations of the heart that are present from birth (congenital heart defects) develop a rare progressive heart condition called Eisenmenger syndrome.
The disorder is characterized by increased blood pressure in the main blood vessel (pulmonary artery) connecting the heart to the lungs (pulmonary hypertension) and improper blood flow within the heart.
The most common congenital heart defect associated with Eisenmenger syndrome is a ventricular septal defect (VSD) or a “hole in the heart”. The “hole” or defect is between the two lower chambers of the heart (left ventricle and right ventricle).
This defect allows blood to flow from the left ventricle into the right ventricle (left-to-right shunt). The shunt causes increased blood flow into the lungs eventually resulting in pulmonary hypertension, which causes progressive damage to the small blood vessels in lungs (pulmonary vascular disease).
As the damage continues, pulmonary hypertension increases and the small blood vessels become thickened or blocked hampering the flow of blood. Ultimately, blood flow is reversed back through the shunt so that blood flows from the right ventricle into the left ventricle (right-to-left shunt) bypassing the lungs completely. A variety of symptoms including life-threatening complications may occur.
Eisenmenger syndrome specifically refers to the combination of pulmonary hypertension and right-to-left shunting of the blood within the heart.
What Are The Symptoms of Pulmonary Hypertension?
The symptoms of Pulmonary Hypertension include shortness of breath, fatigue, syncope (temporary loss of consciousness), chest pain, and leg or ankle swelling. Most of symptoms of pulmonary hypertension result from progressive right ventricular failure.
The right ventricle is responsible for pumping blood through the lungs. With progressive narrowing of the pulmonary arteries, the right ventricle will reach the limit on how much blood it can pump to the lungs and start to fail, developing the symptoms described above. Without treatment, the patient’s condition will steadily worsen and life expectancy will be short.
Treating Pulmonary Hypertension
Medical therapy is the treatment for Pulmonary Arterial Hypertension (PAH).
Treatment may include oxygen, agents to help your heart pump better, diuretics and anticoagulants (blood thinners). Sometimes medications to lower PAH and reverse the scarring in the blood vessels in the lung is given. Lung transplants also are done for PAH, but in congenital heart disease, usually heart and lung transplantation is required.
Medications
Most medications often used to treat coexistent illnesses are safe for people with PAH. Still, care should be taken with prescription medications and over-the-counter drugs. Any anesthetics or sedatives can be very hazardous; ask your physician which medications are safe. Discuss the use of decongestant cold medicines with your doctor.
People with PH and certain underlying congenital heart defects may need antibiotics before certain dental procedures to prevent endocarditis.
Additional Precautions
Additional precautions are often taken with PAH patients. These include supplemental oxygen during air travel, antibiotic therapy for significant respiratory tract infections, pneumococcal pneumonia vaccine and yearly flu vaccines (since pneumonia can be very serious with PH patients).
Also avoid conditions in which the ambient oxygen concentration may be decreased, such as high altitude and travel in unpressurized airplane cabins. Before starting an exercise program, ask your physician what activities are appropriate for you.
Finally, if you’ve begun medical treatment for PAH, stopping any of your medicines without your physician’s approval can be extremely dangerous.
Medical therapy has significantly improved the outlook for most patients with pulmonary hypertension, but it doesn’t “cure” it. Don’t stop medical therapies unless your physician (trained in caring for PAH patients) recommends doing so.
Reference
1. American Heart Association website Updated:Jun 25,2012
| Last Reviewed | : | 15 September 2017 |
| Writer | : | Dr. Ainol Shareha Binti Sahar |
| Translator | : | Dr. Ainol Shareha Binti Sahar |
| Accreditor | : | Dr. Zarina Banu Binti Abdulla |